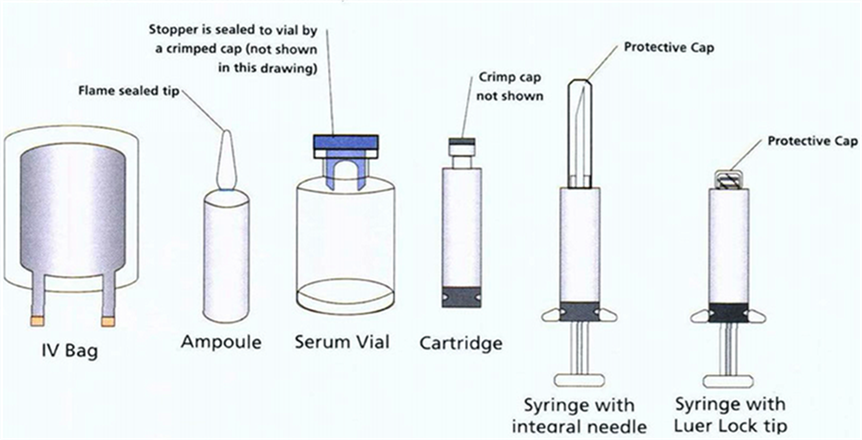
Home > Events >Visual Inspection of Parenteral Products Part 3: Defect Prevention vs Removal QA vs QC
Visual Inspection of Parenteral Products Part 3: Defect Prevention vs Removal QA vs QC
Quality is the degree to which a set of inherent characteristics of an object fulfill requirements ,according to ISO 9001:2015.
Quality
Assurance and Quality Control are separate parts of the Quality Management
System (QMS)
Quality
Assurance
The planned and systematic activities
implemented in a quality system so that required quality standard for a product
or service can be fulfilled.
QA is therefore proactive in its nature and it
is targeted in designing and managing all production processes in such a way to
reduce as much as possible the occurrence of defects and problems in
manufactured products.
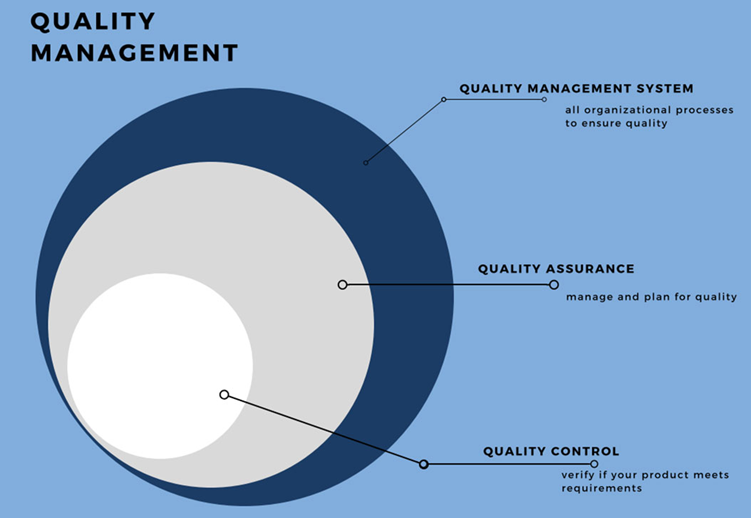
Quality
Control
The procedure or set of procedures intended to
ensure that a manufactured product or performed service adheres to a defined
set of quality criteria or meets the requirements of the client or customer.
QC is therefore reactive in its nature; it is
targeted to find and remove from production stream all products which are not
compliant with quality requirements.
It is extremely important to highlight that
while this document is entirely
management of the whole production and Quality Assurance (QA) process are likewise essential to reduce as much as possible any cause of defective or contaminated products in the first place.

Neither
QA or QC process alone can guarantee 100% good results
They should always work together to help each other to achieve the most effective and stable results on final product quality.

QA and QC – A balance critical to achieve Product Quality
Quality Control in Parenteral
Main inspection/quality items to be checked. After the introduction of QC
and QA concepts we’ll start now diving more in detail about quality control in
parenteral products and which main quality aspects are regulated by main
pharmacopeias and are commonly checked worldwide.
There are obviously important differences and changes in time and among
different countries, however we can list the following most important
categories of visual controls in parenteral products.
·
Visible particles
·
Container Closure Integrity
·
Other visible defects (cosmetic)
·
Additional container defects
We already discussed in part 2 how the most critical aspect to be
controlled before releasing the products to the market and to the final users
is the safety of the product, then it comes its proper function and finally
come minor defects as cosmetic or appearance.
The safety of products can be harmed by two main problems:
- the
presence of foreign particles into the container, especially extrinsic
(see part 2), which by definition implies the lack of sterility
- the
lack of integrity of the container so that microorganisms from outside can
enter and contaminate the product
According to these
principles, the most important visual controls performed on 100% of production
are for particles and closure of container.
The other controls (filling level, cosmetic, etc.) are usually performed at the same time but are less critical and receive less attention and resources.

Regulations, Definitions & Guidelines About Visible
Particles
Starting in 1942 when the USP and the American Pharmaceutical Association (publisher of the National Formulary at that time) stated that aqueous injections should be “substantially free” of particles discernible with the naked eye, the definition eventually evolved into those currently used by the three major Pharmacopeias stating that injectable drug products should be “essentially free” (USP), “practically free” (European Pharmacopeia) or free of “readily detectable” (Japanese Pharmacopeia) visible particles.
Most recent requirements
require each and every unit of injectable product to be inspected as part of
the routine manufacturing process (100% inspection).
It is recommended to
execute inspection where defects can most easily be spotted, for example before
labeling.
Each unit can be examined
manually by unaided eye (Manual Visual Inspection or MVI) or by using a
conveyor system to transport and present the containers to a human inspector
(Semi-automatic inspection) or by means of Automatic Visual Inspection (AVI)
machine.
Manual and semi-automatic
inspection should only be performed by trained, qualified inspectors.
The intent of this inspection is the detection and removal of any observed defect. When in doubt, units should be removed.
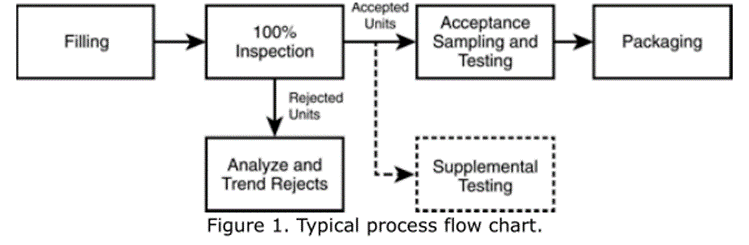
Notes: 100% inspection refers to the complete inspection of the container+closure system and its content.
Requirements
from main Pharmacopeias
- EP
requires “Practically free” of visible particles
- USP
requires “Essentially free” of visible particles
- JP
requires “free from readily detectable foreign insoluble matters”
For a very long time this kind of unclear –
nonobjective requirements have put drug manufacturers in a difficult position.
However recently, the FDA and the other regulatory agencies have moved forward
to make requirements and procedures more standardized, clear and easy to
fulfill and assess.
After several enhancements and harmonization
processes to make all pharmacopeias more similar, the regulation is currently
based on three main steps:
- The
manufacturing processes and associated controls are designed to yield a
product free from visible particles to the extent possible.
- Visual Inspection (100%)
is performed at the end of drug product manufacturing
- Visual
Inspection is followed by sampling inspection (AQL sampling) to confirm
the “Essentially free” requisite before releasing the batch to the market.
Regulations,
definitions & guidelines about Container Closure Integrity (CCI)
Similarly, to requirements for visible particles, the three main pharmacopeias contain general requirements about integrity of closure and are subject to a process of evolution through time and common harmonization.
·
USP “The packaging system should be closed or sealed in
such a manner as to prevent contamination or loss of contents”
· 21 CFR “Container closure systems shall provide adequate protection against foreseeable external factors in storage and use that can cause deterioration or contamination of the drug product”
·
JP “Hermetic containers or tight containers which
are able to prevent microbial contamination are usually used for the
preparations”
·
EP “The tightness of the container is ensured by suitable
means. Closures ensure a good seal, prevent the access of micro-organisms and
other contaminants and usually permit the withdrawal of a part or the whole of
the contents without removal of the closure”
As we will see more in detail in the next part,
the CCI testing can be
nowadays performed by visual inspection and by
other non-visual technologies (high voltage, pressure decay, laser adsorption, etc.).
Consequently, current regulations focus more on
the result and the validation of the whole process regardless of the specific
inspection system/technology used.
It is therefore required to first evaluate and assess the risk of lack of integrity and take every possible measure to prevent its happening (QA approach). Then it is required to take the best possible approach to detect and reject from production all containers whose integrity is not guaranteed(QC approach).